Dialogues Clin Neurosci. 2013 Dec;15(4):431-43.
Abstract
Despite the importance of numerous psychosocial factors, at its core, drug addiction involves a biological process: the ability of repeated exposure to a drug of abuse to induce changes in a vulnerable brain that drive the compulsive seeking and taking of drugs, and loss of control over drug use, that define a state of addiction. Here, we review the types of molecular and cellular adaptations that occur in specific brain regions to mediate addiction-associated behavioral abnormalities. These include alterations in gene expression achieved in part via epigenetic mechanisms, plasticity in the neurophysiological functioning of neurons and synapses, and associated plasticity in neuronal and synaptic morphology mediated in part by altered neurotrophic factor signaling. Each of these types of drug-induced modifications can be viewed as a form of “cellular or molecular memory.” Moreover, it is striking that most addiction-related forms of plasticity are very similar to the types of plasticity that have been associated with more classic forms of “behavioral memory,” perhaps reflecting the finite repertoire of adaptive mechanisms available to neurons when faced with environmental challenges. Finally, addiction-related molecular and cellular adaptations involve most of the same brain regions that mediate more classic forms of memory, consistent with the view that abnormal memories are important drivers of addiction syndromes. The goal of these studies which aim to explicate the molecular and cellular basis of drug addiction is to eventually develop biologically based diagnostic tests, as well as more effective treatments for addiction disorders.
Introduction
Drug addiction, which can be defined as the compulsive seeking and taking of drugs despite horrendous consequences or loss of control over drug use, is caused by long-lasting drug-induced changes that occur in certain brain regions.1 Only some individuals, however, succumb to addiction in the face of repeated drug exposure, while others are capable of using a drug casually and escaping an addiction syndrome. Genetic factors account for roughly 50% of this individual variability in addiction vulnerability, and this degree of heritability holds true for all major classes of addictive drugs, including stimulants, opiates, alcohol, nicotine, and cannabinoids.2 It has not yet been possible to identify most of the genes that comprise this genetic risk, likely due to the involvement of perhaps hundreds of genetic variations summating in a single individual to confer addiction vulnerability (or, in other individuals, resistance).
The other 50% of the risk for addiction is due to a host of environmental factors, occurring throughout a lifetime, that interact with an individual’s genetic composition to render him or her vulnerable to addiction to a greater or lesser extent. Several types of environmental factors have been implicated in addiction, including psychosocial stresses, but by far the most powerful factor is exposure to a drug of abuse itself. Certain “gateway” drugs, in particular, nicotine, have been shown to increase one’s vulnerability to an addiction to another drug.3 Moreover, there is increasing evidence that, despite a range of genetic risks for addiction across the population, exposure to sufficiently high doses of a drug for long periods of time can transform someone who has relatively lower genetic loading into an addict.4
Great progress has been made over the past two decades in identifying both the discrete regions of brain that are important in mediating an addiction syndrome, as well as the types of changes at the molecular and cellular levels that drugs induce in these regions to underlie key aspects of addiction.1,5 The circuit that has received the most attention is referred to as the mesolimbic dopamine system, which involves dopamine neurons in the ventral tegmental area (VTA) of the midbrain innervating medium spiny neurons in the nucleus accumbens (NAc, a part of ventral striatum). These VTA neurons also innervate many other forebrain regions, including hippocampus, amygdala, and prefrontal cortex (PFC).
It makes sense to consider these drug-induced addiction mechanisms in this volume on memory for three overlapping reasons.6
- First, all drug-induced adaptations can be seen as types of “molecular or cellular memory:” the nerve cell undergoing such changes is different as a result of drug exposure and hence responds differently to that same drug, to other drugs, or to a host of other stimuli as a result.
- Second, it is interesting that many, perhaps most, of the types of changes that have been associated with a state of addiction (eg, altered gene transcription, epigenetics, synaptic and whole cell plasticity, and neuronal morphology and neurotrophic mechanisms) are also implicated in traditional forms of “behavioral memory” such as spatial memory, fear conditioning, and operant conditioning, among others.
- Third, among the brain regions affected by drugs of abuse are those that are key neural substrates for behavioral memory, including hippocampus, amygdala, and PFC. This coincides with the increasing realization that some of the most important features of addiction seen clinically (eg, drug craving and relapse) reflect abnormalities in traditional memory circuits, with long-term memories of the drug experience serving as potent drivers of addiction pathology.4,7,8 Conversely, the brain’s reward regions (eg, VTA and NAc) potently influence behavioral memory.
This article provides an overview of the major types of molecular and cellular changes that occur in several brain regions in animal models of addiction, concentrating on the nucleus accumbens for which most information is currently available. Importantly, it has been possible increasingly to validate some of these changes in human addicts based on studies of postmortem brains. Despite the fact that drugs of abuse have distinct chemical structures and act on distinct protein targets, it is striking that many prominent addiction-related adaptations are common to many, and in some cases all, drugs of abuse and likely contribute to shared features of an addiction syndrome.4,9 In contrast, many other drug-induced adaptations are specific to a given drug and may mediate more unique aspects of a given addiction. We focus here on stimulant and opiate drugs of abuse, which produce more dramatic effects in animal models compared with other drugs. We also highlight important areas for future research that will further increase our knowledge of addiction syndromes and translate these advances into improved diagnostic tests and treatments.
Transcriptional and epigenetic mechanisms
The knowledge that addicts can remain at increased risk for relapse despite years of abstinence means that addiction involves drug-induced changes in the brain that can be very stable. This has led several groups to consider changes in gene expression as an important component of the addiction process (Figure 1). Accordingly, studies of candidate genes or genome-wide investigations involving DNA microarrays and more recently RNA-seq (high-throughput sequencing of expressed RNAs) has identified numerous genes whose expression is altered in a given brain region in rodent and primate models of addiction and in human addicts (eg, refs 10-17). Examples of such genes are discussed in subsequent sections of this review.
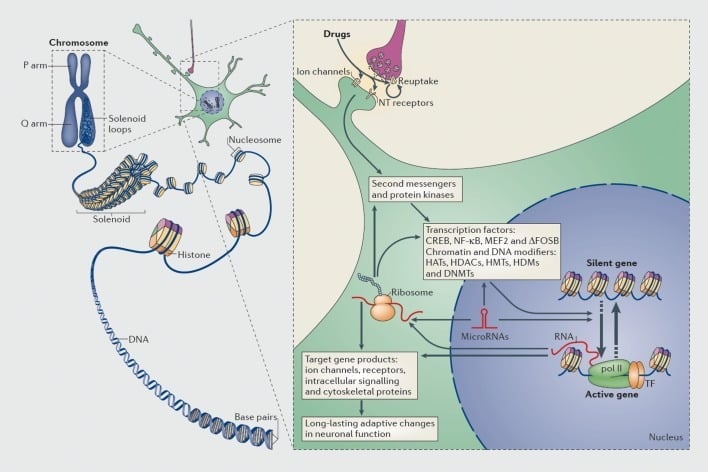
Mechanisms of transcriptional and epigenetic regulation by drugs of abuse. In eukaryotic cells, DNA is organized by wrapping around histone octomers to form nucleosomes, which are then further organized and condensed to form chromosomes (left part). Only by temporarily unraveling compacted chromatin can the DNA of a specific gene be made accessible to the transcriptional machinery. Drugs of abuse act through synaptic targets such as reuptake mechanisms, ion channels, and neurotransmitter (NT) receptors to alter intracellular signaling cascades (right part). This leads to the activation or inhibition of transcription factors (TFs) and of many other nuclear targets, including chromatin-regulatory proteins (shown by thick arrows); the detailed mechanisms involved in the synaptic regulation of chromatin-regulatory proteins remain poorly understood. These processes ultimately result in the induction or repression of particular genes, including those for noncoding RNAs such as microRNAs; altered expression of some of these genes can in turn further regulate gene transcription. It is proposed that some of these drug-induced changes at the chromatin level are extremely stable and thereby underlie the long-lasting behaviours that define addiction. CREB, cyclic AMP-responsive element binding protein; DNMTs, DNA methyltransferases; HATs, histone acetyltransferases; HDACs, histone deacetylases; HDMs, histone demethylases; HMTs, histone methyltransferases; MEF2, myocyte-specific enhancer factor 2; NF-kB, nuclear factor-KB; pol II, RNA polymerase II. Reproduced from ref 44: Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011 ;12:623-637.
Likewise, many types of transcription factors—proteins that bind to regulatory regions of genes and thereby increase or decrease the transcription of those genes—have been implicated in mediating the long-term effects of drug of abuse on gene expression in the brain. Prominent examples include CREB (cAMP response element binding protein), ΔFosB (a Fos family transcription factor), NFkB (nuclear factor kB), MEF2 (myocyte enhancing factor-2), and glucocorticoid receptors, among several others.5,10,18–22 It has been increasingly possible to understand the cellular signaling pathways through which drugs of abuse activate a given transcription factor in brain and to causally link such activation to that transcription factor’s target genes and to specific behavioral aspects of addiction (see Figure 1). This progress is illustrated by consideration of CREB and ΔFosB, which are the best studied transcription factors in addiction models.
cAMP Response element binding protein
Stimulant and opiate drugs of abuse activate CREB in several brain regions important for addiction, including prominently in the NAc.23,24 CREB is known to be activated in other systems by cAMP, Ca2+, and growth factor pathways,25 and it is not yet known which of these mediates its activation in NAc by drugs of abuse. Drug activation of CREB in NAc has been shown to represent a classic negative feedback mechanism, whereby CREB serves to reduce an animal’s sensitivity to the rewarding effects of these drugs (tolerance) and to mediate a negative emotional state during drug withdrawal (dependence).18,26,27 These effects have been shown recently to drive increased drug self-administration and relapse, presumably through a process of negative reinforcement.28 These actions of CREB seem to involve both major subtypes of NAc medium spiny neurons, those that express predominantly D1 versus D2 dopamine receptors.24 Interestingly, a large body of literature has shown that CREB, acting in hippocampus and amygdala, is a key molecule in behavioral memory.29–31 This broad role in addiction and behavioral memory likely reflects the fact that neurons are imbued with a finite number of molecular mechanisms with which to adapt to a constantly changing environment.
Target genes for CREB that mediate this behavioral phenotype have been identified through genome-wide assays as well as more selected efforts.10,18,32 One example is the opioid peptide dynorphin: stimulant induction of dynorphin expression in NAc neurons, mediated via CREB, increases dynorphin activation of k opioid receptors on VTA dopamine neurons and thereby suppresses dopaminergic transmission to the NAc and impairs reward.18 Several other CREB targets have been shown to be important for drug-induced synaptic plasticity, as discussed below. While CREB is also activated in several other brain regions by stimulants and opiates,23,24 less is known about the behavioral consequences of this effect and the target genes through which they occur. Likewise, less is known about CREB’s role in mediating the actions of other drugs of abuse.19
ΔFosB
Acute exposure to virtually any drug of abuse induces all Fos family transcription factors in NAc and several other brain regions. This induction is rapid but also highly transient, with Fos protein levels reverting to normal within 8 to 12 hours. Uniquely among these Fos family proteins is ΔFosB, a truncated product of the FosB gene, which by virtue of its unusual stability, gradually accumulates through a course of repeated drug exposure and becomes the predominant Fos protein expressed under these conditions.22,33 Moreover, because of this stability, levels of ΔFosB persist for weeks after drug withdrawal. Such chronic induction of ΔFosB has been demonstrated for virtually all drugs of abuse34 and, for most drugs, is selective for Dl-type NAc neurons.34,35 It has also been demonstrated in human addicts.35 A large body of literature has demonstrated that such ΔFosB induction in D1-type NAc neurons increases an animal’s sensitivity to drug as well as natural rewards and promotes drug self-administration, presumably through a process of positive reinforcement (see refs 34 to 38). Interestingly, drug induction of ΔFosB in NAc is more dramatic in adolescent animals, a time of greater addiction vulnerability,39 and its induction by nicotine has been shown to mediate nicotine’s gateway-like enhancement of cocaine reward.40
As for CREB, numerous target genes for ΔFosB have been identified in NAc by use of candidate gene and genome-wide approaches.10,32 While CREB induces dynorphin, ΔFosB suppresses it, which contributes to ΔFosB’s pro-reward effects.38 Another ΔFosB target is cFos: as ΔFosB accumulates with repeated drug exposure it represses c-Fos and contributes to the molecular switch whereby ΔFosB is selectively induced in the chronic drug-treated state.41 Many other ΔFosB targets have been shown to mediate the ability of certain drugs of abuse to induce synaptic plasticity in the NAc and associated changes in the dendritic arborization of NAc medium spiny neurons, as will be discussed below.
The functional consequences of ΔFosB induction in other brain regions is less well understood, although its induction in orbitofrontal cortex (OFC) has been studied in some detail. Here, ΔFosB mediates tolerance that occurs to the cognitive-disrupting effects of cocaine during a course of chronic exposure, and this adaptation is associated with increased cocaine self-administration.42,43
Genome-wide assays have suggested several potential target genes that mediate these effects.42 Despite ΔFosB’s unique temporal properties, and the knowledge that it is induced in traditional memory circuits (eg, hippocampus), there has not yet been an exploration of the role of ΔFosB in behavioral memory, an interesting subject for future research.
Epigenetic mechanisms
In more recent years, studies of transcription have been pushed one step further to epigenetics44 (see Figure 1), which can be broadly defined as a change in gene expression that occurs in the absence of a change in DNA sequence. Epigenetic mechanisms control the packaging of DNA within a cell nucleus via its interactions with histones and many other types of nuclear proteins, which together comprise chromatin. Gene expression is controlled by the state of this packaging through the covalent modification of histones, other proteins, and DNA itself. As just some examples, acetylation of histones tends to promote gene activation, methylation of histones can either promote gene activation or repression depending on the Lys residue undergoing this modification, and methylation of DNA is generally associated with gene repression although certain variant forms of methylation (eg, 5-hydroxymethylation) may be associated with gene activation.
Epigenetics is an appealing mechanism because, in other systems, for example, developmental and cancer biology, certain epigenetic modifications can be permanent. For this reason, epigenetics has been pursued both in learning and memory models (eg, refs 45-48) as well as in addiction;44,49 in both systems profound changes have been reported in histone acetylation and methylation and in DNA methylation. As just one example, the histone methyltransferase, G9a, is implicated in both memory50 and addiction.51,52 In addiction models, G9a expression is downregulated in NAc in response to stimulant or opiate drugs of abuse and this has been shown to enhance the rewarding effects of these drugs.51,52 Interestingly, cocaine suppression of G9a is mediated by ΔFosB. G9a catalyzes the dimethylation of Lys9 of histone H3 (H3K9me2), a major mediator of gene repression. ChIP-chip or ChIP-seq (chromatin immunoprecipitation followed, respectively, by promoter chips or high-throughput sequencing) has been used to obtain genome-wide maps of the genes in NAc that display altered H3K9me2 after stimulant or opiate exposure.32,52,53 By overlapping these gene lists with genome-wide lists of gene expression changes, and with genome-wide maps of many other forms of epigenetic modifications (eg, ΔFosB binding, CREB binding, other histone modifications, etc),32,53 it should be possible to identify an increasingly complete set of genes that are regulated by drugs of abuse and to understand the underlying epigenetic mechanisms involved.
Another form of epigenetic regulation implicated in memory and addiction is the generation of microRNAs. These small, noncoding RNAs bind to complementary regions of mRNAs and thereby suppress their translation or induce their degradation. Deletion of Argonaut, a protein crucial for the processing of miRNAs, alters behavioral responses to cocaine, with distinct effects observed for D1- versus D2-type medium spiny neurons.54 Several specific miRNAs have likewise been shown to be regulated by drug exposure and, in turn, to influence behavioral responses to the drugs (eg, refs 55,56). It will be exciting in future studies to identify the mRNA targets of these miRNAs and characterize how they affect the addiction process.
Synaptic plasticity
The same general types of synaptic modifications at glutamatergic synapses, which have been implicated in hippocampus and amygdala in behavioral memory (see other articles in this issue), have similarly been shown to occur in brain reward regions in addiction models and to be important in mediating the addiction process.57,58 Such drug-induced synaptic plasticity has been described in several brain regions, however, we concentrate here on NAc where most of the research has focused to date (Figure 2).
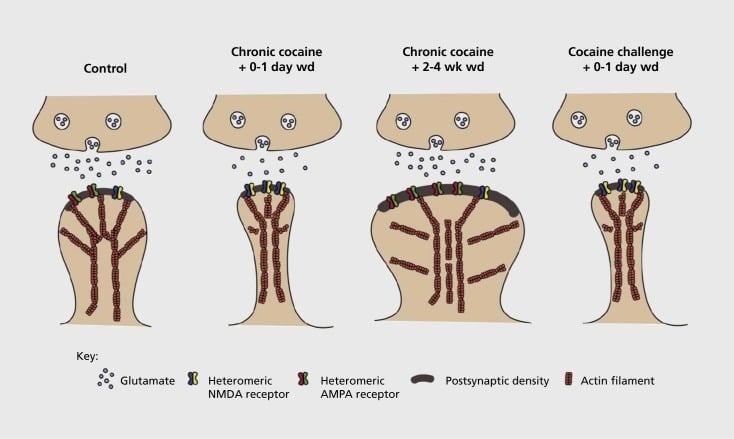
Model of addiction-related synaptic and structural plasticity in nucleus accumbens (NAc). Chronic exposure to cocaine results in a time-dependent and transient reorganization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartic acid (NMDA) glutamate receptors at NAc medium spiny neuron (MSN) synapses, as well as structural changes in the spine head of NAc MSNs that correlate with distinct forms of synaptic plasticity. For example, chronic cocaine induces surface expression of NMDA receptors, silent synapse formation and long-term depression (LTD) at early withdrawal time points. During more prolonged withdrawal (wd), these synaptic changes reverse with the result being increased expression of surface AMPA receptors, a consolidation of the synapse into a mushroom-shaped spine and long-term potentiation (LTP). These effects rapidly revert back again upon exposure to a challenge dose of cocaine leading to restructuring of the spine into thin spines and a depression of synaptic strength.
Initial experiments demonstrated that repeated exposure to stimulant drugs of abuse induces an LTD (long-term depression)-like state at glutamatergic synapses in the NAc.59 However, more recent work has demonstrated such plasticity to be highly time-dependent, with LTD occurring early after the last cocaine exposure evolving into more of an LTP (long-term potentiation)-like state after longer withdrawal time points.60,61 This work, which to date has been carried out primarily using investigator administered—as opposed to self-administered—drugs, has defined the need for more systematic investigations in self-administration models that track the forms of synaptic plasticity that occur at glutamatergic synapses in NAc over a detailed time course from acquisition of self administration to its maintenance, through different times of withdrawal and extinction, and in response to relapse-evoking stimuli. Work to date has also defined some of the molecular mechanisms that contribute to this drug-induced synaptic plasticity, including the trafficking of AMPA receptors to the synapse perhaps mediated in part via CaMKII (Ca2+/calmodulin-dependent protein kinase II) phosphorylation of certain AMPA receptor subunits as well as altered expression of AMPA receptor subunits (eg, 60,62-65, Figures 2 and 3). A role for CREB and ΔFosB has been implicated in these phenomena, as well as in associated changes in the morphology of glutamatergic synapses (see below). For example, GluAl is a target for CREB in NAc, where GluA2 and CaMKII are both targets of ΔFosB, in this brain region .35,36,66,67 Moving forward, it will be important to link specific adaptations to time-dependent changes in synaptic function and behavioral features of addiction.
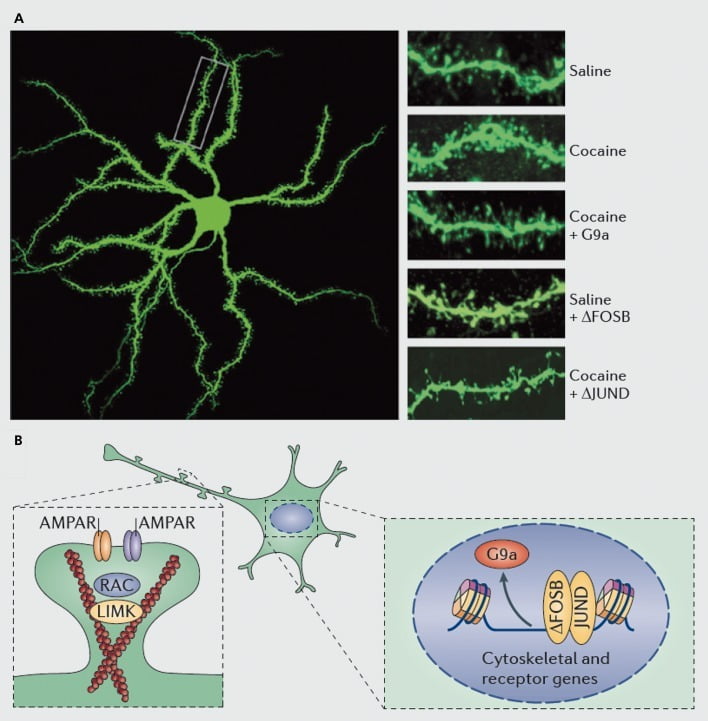
Molecular mechanisms underlying cocaine induction of dendritic spines on nucleus accumbens (NAc) medium spiny neurons. A) shows cocaine-induced increases in dendritic spine number that can be blocked by viral overexpression of G9a or JunD (an antagonist of AP1 -mediated transcription), or mimicked by viral overexpression of FosB. B) Regulation of AMPA receptor (AMPAR) trafficking and of the actin cytoskeleton (left), as well as regulation of the transcription of glutamate receptors and actin regulatory proteins (eg, as mediated via ΔFosB, right) have been shown to play important roles in mediating cocaine’s regulation of NAc dendritic spine density. UMK, LIM domain kinase; RAC, Ras-related C3 botulinum toxin substrate.
New experimental tools are making it possible for the first time to define with increasing precision which particular circuits display these forms of synaptic plasticity and what behavioral abnormalities they mediate. For example, the shell and core subregions of NAc display differences in drug-induced synaptic plasticity, as do D1- versus D2-type medium spiny neurons within each subregion.60,63,64,67 Likewise, optogenetic experiments have provided novel insight into the contribution of a particular form of synaptic plasticity (eg, LTD) at specific populations of glutamatergic synapses in NAc, for example, those arising from medial PFC versus basolateral amygdala versus ventral subiculum (the major output of hippocampus).68–70 Ultimately, it will be necessary to overlay drug-induced molecular adaptations in each of these afferent neurons with synapse-specific adaptations that occur in their postsynaptic dendrites to compile a complete understanding of how drugs of abuse modify the brain’s circuitry to drive particular aspects of the addicted state. This endeavor will require a greater appreciation of drug-induced plasticity at inhibitory synapses within these same brain regions, an area that has received very little attention to date.65
Whole cell plasticity
While the majority of research involving neurophysiological changes in neurons in drug abuse phenomena, as in learning and memory phenomena, has focused on synaptic plasticity, there is increasing evidence for the importance of whole cell plasticity as well. Whole cell plasticity, also referred to as homeostatic plasticity,71 involves changes in the intrinsic excitability of an entire nerve cell in a manner that it is not synapse-specific. Given that certain features of drug addiction involve enhanced or reduced sensitivity to a drug, it makes sense that enhanced or reduced electrical excitability of certain nerve cells contributes to these behavioral adaptations.5
The best established example of whole cell plasticity to a drug of abuse is the ability of chronic opiates to increase the intrinsic excitability of noradrenergic neurons of the locus coeruleus (LC).72 This increased excitability is mediated via CREB and its induction of certain isoforms of adenylyl cyclase, which drive increased firing of LC neurons perhaps through the induction of Na+ channels.72–75 This hyperexcitabilty of LC neurons represents a classic mechanism of tolerance and dependence and drives some of the signs and symptoms of opiate withdrawal. Interestingly, CREB mediates a similar form of whole cell plasticity in NAc medium spiny neurons, which are also rendered hyperexcitable by chronic exposure to drugs of abuse via CREB.76 It will thus be critical in future investigations to understand how CREB-mediated synaptic plasticity of glutamatergic synapses on NAc medium spiny neurons65,66 summates with CREB-mediated intrinsic hyperexcitability of these neurons76 to control behavioral features of addiction.
Another example of whole cell plasticity in addiction models is the hyperexcitability of VTA dopamine neurons that occurs after chronic exposure to opiate drugs of abuse (Figure 4).77,78 This adaptation, which has been linked to morphological changes in these nerve cells (see next section), is not mediated by CREB but achieved instead via regulation of neurotrophic signaling cascades, as described below.

Working model of chronic morphine-induced adaptations in ventral tegmental area (VTA) dopamine neurons. Chronic morphine decreases VTA dopamine (DA) soma size yet increases neuronal excitability, while dopamine transmission to the nucleus accumbens is decreased. The net effect of morphine is a less responsive reward pathway, ie, reward tolerance. Downregulation of IRS2-AKT signaling in VTA mediates the effects of chronic morphine on soma size and electrical excitability; the effect on excitability is mediated via decreased γ-aminobutyric acid (GABA)A currents and suppression of K’ channel expression. Morphine-induced downregulation of mTORC2 activity in VTA is crucial for these morphine-induced morphological and physiological adaptations as well as for reward tolerance. In contrast to mT0RC2, chronic morphine increases mTORCI activity, which does not influence these morphine-induced adaptations. BDNF, brain-derived neurotrophic factor; IRS, insulin receptor substance; mTORC, mTOR complex; AKT, protein kinase B Reproduced from ref 77
Morphological plasticity and neurotrophic mechanisms
Increasing evidence, much of it from studies of hippocampal and cerebral cortical neurons, has shown that changes in synaptic plasticity are associated with morphological changes at synapses. For example, LTD and the generation of silent synapses are associated with the formation of thin or stubby dendritic spines, whereas LTP is associated with larger, mushroom-shaped spines.79,80 It is thus interesting that the drug abuse field has focused on drug-induced changes in dendritic spines for >15 years. Chronic exposure to stimulant drugs of abuse increases the dendritic spine density of medium spiny neurons of the NAc, a change that predominates for Dl-type neurons.67,81,82 Induction of spines has been associated for the most part with sensitized behavioral responses to these drugs, although some evidence conflicts with this view.
As with studies of synaptic plasticity, however, far more work is needed to systemically define the changes in dendritic spines that occur during a course of drug self-administration, withdrawal, and relapse. Studies to date, involving investigator- and self-administered drug, suggest very different spine changes occurring at different withdrawal time points and in NAc shell versus core subregions.83–86 It will also be important to define the precise molecular mechanisms by which cocaine or another stimulant produces these time-dependent and cell-type specific effects. ΔFosB has been shown to be both necessary and sufficient for the induction of immature spines on Dl-type NAc neurons.35,51,67 Such regulation occurs in concert with cocaine and ΔFosB regulation of several proteins known to control the reorganization of the actin cytoskeleton. As just one example, transcriptional regulation of several guanine nucleotide exchange factors and GTPase activating proteins poises Rac1, a small GTPase, for transient decreases in activity in response to each cocaine exposure, and such pulsatile decreases in Rac1 activity have been shown, using optogenetic control of Rac1, to mediate induction of immature spines.87 These effects of Racl presumably occur through its control of cofilin and other actin regulatory proteins, which have also been shown to mediate cocaine regulation of spine growth.87,88 However, it is important to emphasize that this is just one pathway involved in cocaine’s regulation of immature spines, since several other proteins have been shown to play an essential role as well, including CDK5 (cyclin-dependent kinase-5), CaMKII, NFkB, MEF2, CREB, G9a, and DNMT3 (DNA methyltransf erase 3a), to name a few.20,21,35,51,67,89,90 Interestingly, cocaine regulation of several of these genes, including induction of CDK5, CaMKII, and NFkB, and repression of G9a, is also mediated via ΔFosB.20,35,51,91
Surprisingly, opiate drugs of abuse exert the opposite effect and reduce dendritic spine density of NAc medium spiny neurons.81 Little is known about the behavioral consequences of this adaptation and the underlying molecular mechanisms involved. This phenomenon is, however, surprising, given that CREB and ΔFosB are induced by both stimulants and opiates and are both implicated in stimulant-mediated induction of NAc dendritic spine density. This raises the question of how opiates suppress NAc spine density despite their induction of these factors.
The other major form of morphological plasticity seen in drug abuse models is the physical reduction in cell soma size of VTA dopamine neurons induced by chronic opiate administration.77,92,93 A similar adaptation occurs in response to cannabinoids.94 This shrinkage of VTA dopamine neurons, which occurs with opiate self- administration93 and has been documented in human heroin addicts examined postmortem,77 seems to mediate reward tolerance and is associated with reduced dopamine release in the NAc. Considerable evidence now indicates that this reduction in cell soma size is mediated by opiate suppression of brain-derived neurotrophic factor (BDNF) expression within these neurons. We have directly linked this opiate-induced withdrawal of BDNF support, and VTA neuron shrinkage, to reduced activity of downstream BDNF signaling cascades in VTA dopamine neurons, specifically reduced activity of IRS2 (insulin receptor substrate-2), AKT (a serine-threonine kinase), and TORC2 (target of rapamycin-2, which is insensitive to rapamycin).77,93 We have also linked this downregulation of BDNF signaling directly to the increased excitability that morphine induces in these neurons, as noted earlier.77,78 Indeed, the decreased cell soma size and increased excitability are tightly coupled, as induction of one leads to the other and vice versa. This control over cell excitability involves suppression of K+ channels and of GABAA current in these neurons.
This role for BDNF in controlling morphine responses at the level of the VTA contrasts with its very different involvement in the actions of cocaine and other stimulants. Stimulants induce BDNF signaling to the NAc, an effect due to increased local synthesis of BDNF as well as increased release from several afferent regions.95 Moreover, increased BDNF signaling in NAc, but not in the VTA, has been shown to promote the behavioral effects of these drugs including their self-administration.95,96 The opposite regulation of BDNF signaling in the VTA-NAc pathway by opiates versus stimulants raises the possibility that such differences mediate the drugs’ opposite regulation of NAc dendritic spines, a possibility now under investigation.
Future directions
The above narrative underscores the tremendous advances that have been made in understanding the molecular and cellular adaptations that occur in brain reward regions in response to repeated exposure to a drug of abuse, and in relating individual adaptations to certain behavioral features of addiction syndromes in animal models. Despite these advances, major questions remain. Most of our existing knowledge focuses on the VTA and NAc, with much less information available about other key limbic brain regions that are also crucial for drug addiction. In addition, all experimental demonstrations of the causal role of a molecular-cellular adaptation in a drug-related behavior have manipulated individual adaptations one at a time. To manipulate numerous adaptations at the same time is clearly far more difficult, but it is also essential, since we know that drugs produce a large number of disparate types of changes even within individual neurons, which likely summate in complicated ways to influence behavior. Such a systems biology approach will be crucial to ultimately cracking the biological underpinnings of addiction. Finally, efforts to understand the molecular-cellular mechanisms of memories related to addiction find themselves at the point at which all other efforts to understand the biological basis of behavioral memory now struggle: our ability to relate biological phenomena to complex behavioral memory remains extremely difficult. Overcoming this divide represents perhaps the greatest challenge in the neurosciences.
Selected abbreviations and acronyms
- Nac
- nucleus accumbens
- CREB
- cAMP response element binding protein
- ΔFosB
- a Fos family transcription factor
- VTA
- ventral tegmental area
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- LTD
- long-term depression
- LTP
- long-term protentiation
- BDNF
- brain-derived neurotrophic factor
- NKkB
- nuclear factor kB
REFERENCES